2020, 47(2): 69-83.
doi: 10.1016/j.jgg.2019.11.009
Abstract:
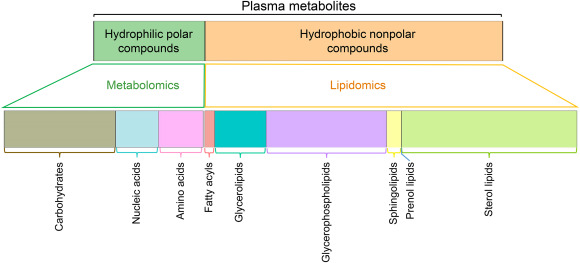
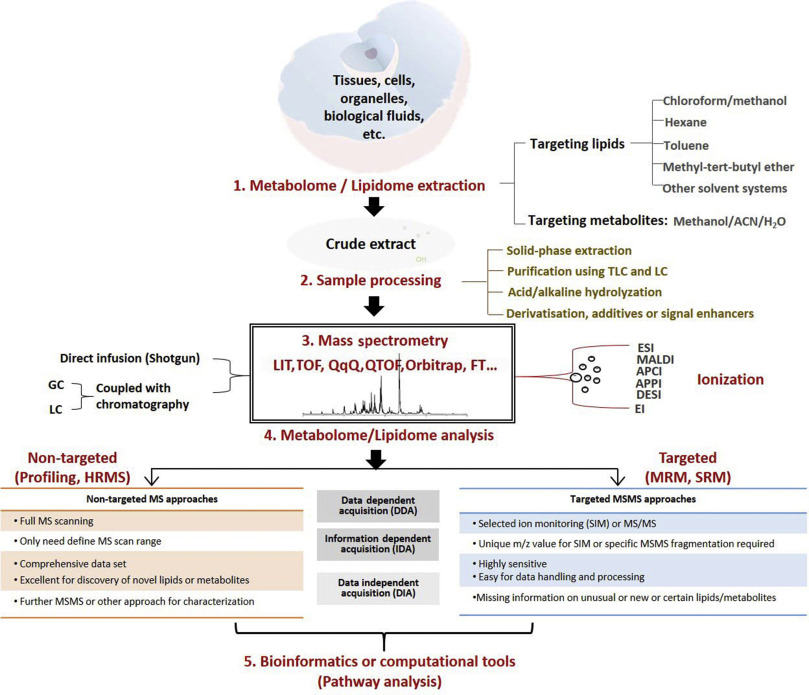
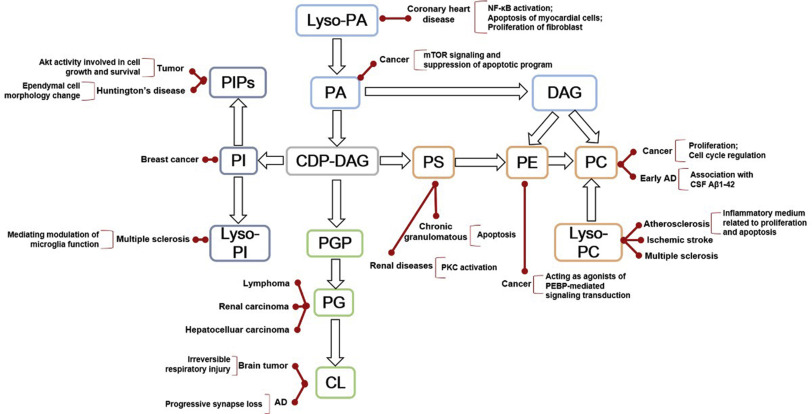
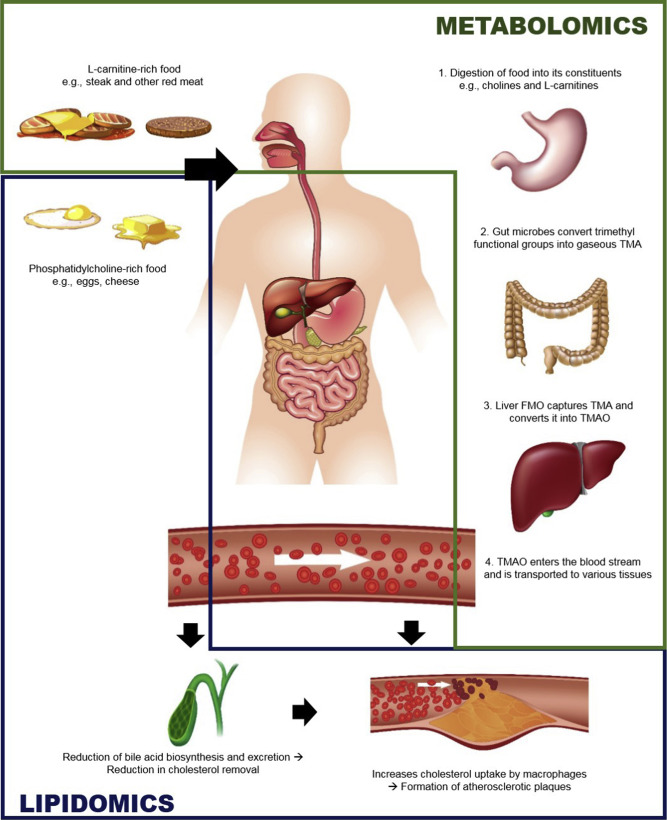
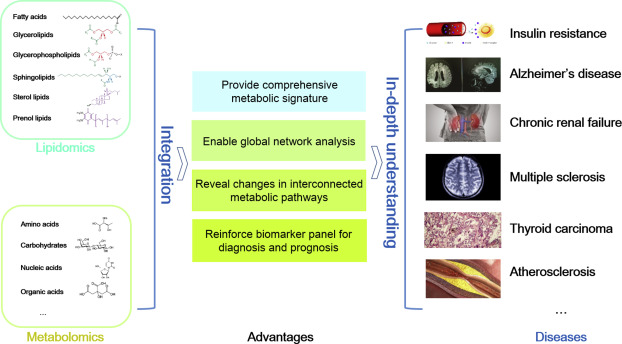
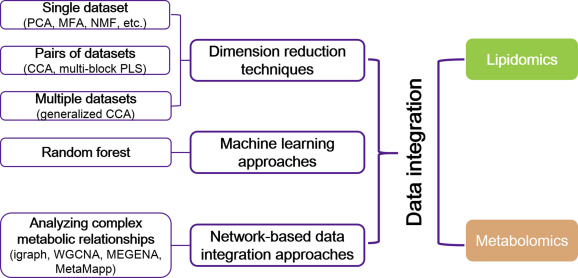
Mass spectrometry (MS)-based omics technologies are now widely used to profile small molecules in multiple matrices to confer comprehensive snapshots of cellular metabolic phenotypes. The metabolomes of cells, tissues, and organisms comprise a variety of molecules including lipids, amino acids, sugars, organic acids, and so on. Metabolomics mainly focus on the hydrophilic classes, while lipidomics has emerged as an independent omics owing to the complexities of the organismal lipidomes. The potential roles of lipids and small metabolites in disease pathogenesis have been widely investigated in various human diseases, but system-level understanding is largely lacking, which could be partly attributed to the insufficiency in terms of metabolite coverage and quantitation accuracy in current analytical technologies. While scientists are continuously striving to develop high-coverage omics approaches, integration of metabolomics and lipidomics is becoming an emerging approach to mechanistic investigation. Integration of metabolome and lipidome offers a complete atlas of the metabolic landscape, enabling comprehensive network analysis to identify critical metabolic drivers in disease pathology, facilitating the study of interconnection between lipids and other metabolites in disease progression. In this review, we summarize omics-based findings on the roles of lipids and metabolites in the pathogenesis of selected major diseases threatening public health. We also discuss the advantages of integrating lipidomics and metabolomics for in-depth understanding of molecular mechanism in disease pathogenesis.